Putting it all together: analysis workflows
For a subset of use cases, we have outlined how specific analysis functionalities could be assembled into analysis workflows to obtain required results from the initial genetic or genomic data. It is worth noting that there may be several viable ways to obtain a given result, based on the features of the Plasmodium genetic data. Within a given analysis workflow path several available tools might interchangably provide a given functionality as output. As both analytical tools and analysis approaches mature, some workflows may not be sequential if multiple parameter values can be estimated jointly (eg COI, phase and frequencies).
Monitoring the prevalence/frequency of drug or diagnostic resistance markers
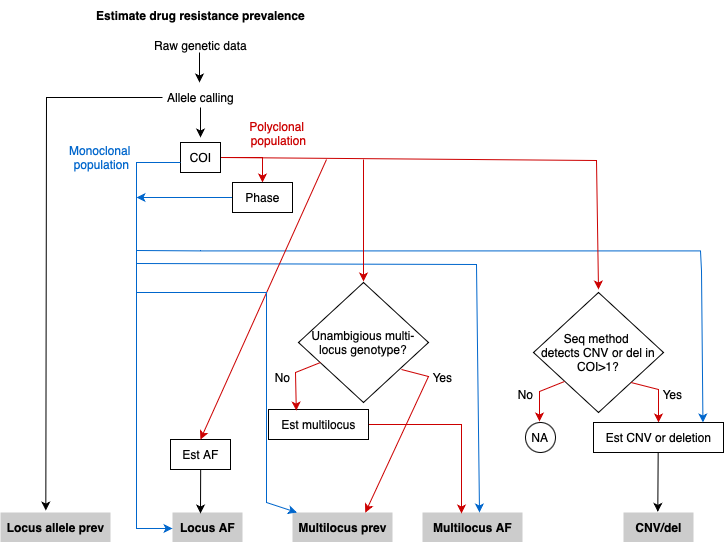
To estimate drug resistance marker prevalence, raw genetic data is processed through allele calling and subsequent steps vary depending on the complexity of infection (COI) and whether the population is monoclonal or polyclonal (including monoclonal and/or polyclonal infections). If the population is monoclonal, allele prevalence (prev) and allele frequencies (AF) at a single locus can be estimated directly and will be identical. Additionally, multi-locus prev, AF and copy number variation (CNV) or deletions (del) can be estimated directly if relevant. If the population is polyclonal and phasing is possible, the analysis follows the same pathway as for a monoclonal population. If phasing is not possible or not performed, additional steps are required depending on whether unambiguous multilocus genotypes can be obtained and whether the sequencing (seq) method utilized detects CNV or deletions in polyclonal infections.
Estimating transmission intensity
![]()
To estimate transmission intensity, raw genetic data is processed through allele calling, complexity of infection (COI) and allele frequency (AF) estimation. Subsequent metric estimation involves measuring the proportion of polyclonal infections in the population, between and within-host relatedness and population diversity, which can all be collectively translated into estimates of transmission. Alternative analysis pathways may be used depending on whether phasing is possible or performed.
Classifying malaria cases as locally acquired or imported from another population
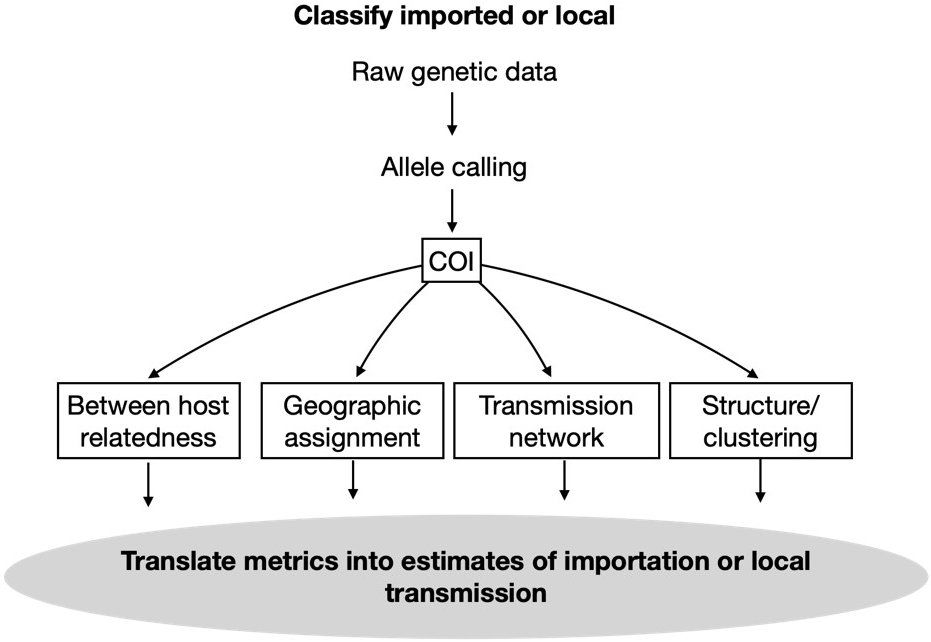
To classify cases as imported or locally acquired raw genetic data is processed through allele calling, followed by estimation of complexity of infection (COI). Then estimation of between host relatedness, geographic assignment, transmission networks and structure and/or clustering metrics can be translated into estimates of importation or local transmission.