Epidemiological Parameters: Ebola Bundibugyo Virus (BVD)
Based on the Epireview database — Nash et al. (2024)
A.Vicco, R. McCabe, T. Naidoo, R.K. Nash, S. Bhatia, H.J.T. Unwin, A. Cori
on behalf of the PERG group
29/05/2026
Source:vignettes/ebola_vignette_BVD.Rmd
ebola_vignette_BVD.RmdDOI: https://doi.org/10.5281/zenodo.20443136
Background
Ebola Bundibugyo virus (BVD) is one of six known Ebolavirus species. It was first identified during the 2007–2008 outbreak in Bundibugyo District, Uganda. This report summarises epidemiological parameters extracted from the Epireview structured database (Nash et al., 2024).
The dataset includes epidemiological parameter estimates (reproduction number, human delays, mutations, risk factors) and information on population settings.
Parameters included
| Category | Parameter types |
|---|---|
| Transmission | R₀, Rₑ ,Attack rate |
| Natural history | Incubation period, serial interval, delays |
| Severity | CFR |
| Genomics | Substitution rate |
| Epidemiology | Risk factors |
Note: Risk factors and Rₑ are reported in tables only, as no extractable numeric values were available. Please refer to the original paper for further details.
Results
How to navigate: Use the tabs below to switch between the main figure and the parameter tables. Click Figure to view the parameter plot, or select any of the Table tabs to explore, filter, and export the underlying data. Click Previous and Next to explore all tables entries. Click on CSV or Excel to download the relative data or copy the data by clicking on Copy.
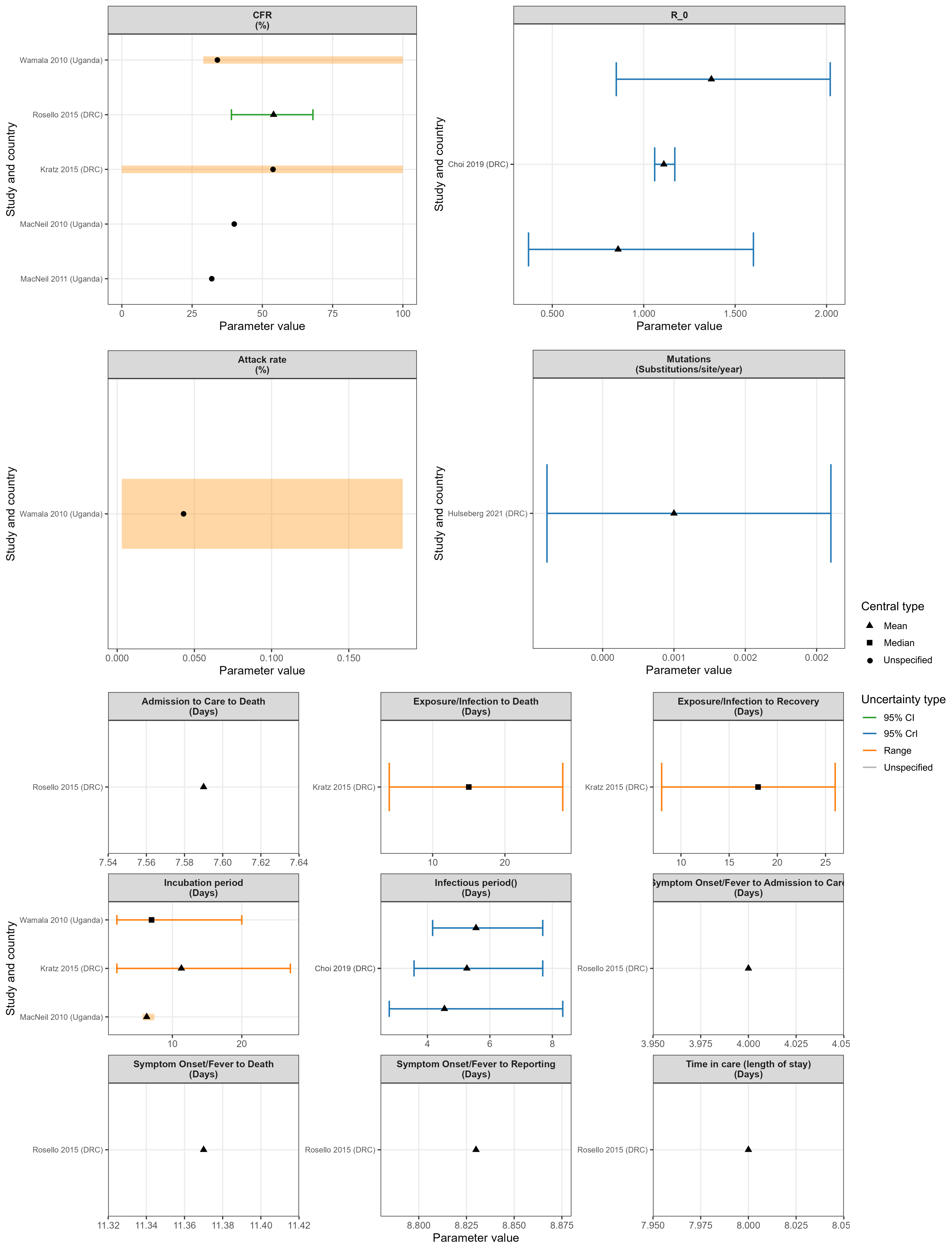
Figure 1. Epidemiological parameters for Ebola Bundibugyo virus. Points represent central estimates (shape = value type); error bars show uncertainty intervals (colour = uncertainty type); orange shading indicates reported parameter ranges.
Methods
Data were retrieved from the Epireview R package
(mrc-ide/epireview). Parameters were filtered to retain
only those attributed to the Bundibugyo species.
Inverse parameters were back-transformed and exponent corrections applied where indicated in the database fields. Uncertainty bounds were combined into a single interval string for display purposes.
Risk factor rows and Rₑ estimates (where no numeric value was extractable) were excluded from figures but retained in Table 2 and Table 3 respectively.
Appendix: Code to Reproduce the Analysis
How to navigate: Use the tabs below to browse the code by section. Click each tab to expand the corresponding script, from data loading and processing through to table generation and plot production. All code chunks can also be individually expanded or collapsed using the Code buttons on the right-hand side of each block.
# Uncomment to install the package from GitHub:
# remotes::install_github("mrc-ide/epireview")
library(tidyverse)
library(epireview)
library(ggplot2)
library(DT)
articles <- epireview::load_epidata_raw("ebola", "article")
models <- epireview::load_epidata_raw("ebola", "model")
params <- epireview::load_epidata_raw("ebola", "parameter")
# Assign QA scores and attach to parameters
articles <- epireview::assign_qa_score(articles = articles)$articles
qa_scores <- articles %>% dplyr::select(covidence_id, qa_score)
params <- params %>% left_join(qa_scores)
# Overview of species in the dataset
# unique(params$ebola_species)
# Join parameters with article-level metadata
df <- left_join(
params,
articles[, c("covidence_id", "first_author_surname", "year_publication",
"article_label", "doi", "notes")],
by = "covidence_id"
) %>%
arrange(article_label, -year_publication)
# Filter for Bundibugyo species only
BVD <- filter(df, ebola_species == "Bundibugyo")
# Identify numeric uncertainty/value columns
unc_cols <- names(BVD)[
grepl("uncertainty|upper|lower|value", names(BVD)) &
sapply(BVD, is.numeric)
]
BVD <- BVD %>%
# Back-transform inverse parameters
mutate(
across(
all_of(unc_cols),
~ if_else(
inverse_param == TRUE & !is.na(.x) & .x != 0,
1 / .x,
.x
)
)
) %>%
# Apply exponent correction
mutate(
across(
all_of(unc_cols),
~ if_else(
!is.na(exponent) & exponent != 0,
.x * (10 ^ exponent),
.x
)
)
) %>%
# Round values and construct combined uncertainty/range strings
mutate(
parameter_value = round(parameter_value, 3),
parameter_uncertainty_single_value = round(parameter_uncertainty_single_value, 3),
comb_uncertainty = if_else(
inverse_param == TRUE,
paste0(
round(parameter_uncertainty_upper_value, 3),
" - ",
round(parameter_uncertainty_lower_value, 3)
),
paste0(
round(parameter_uncertainty_lower_value, 3),
" - ",
round(parameter_uncertainty_upper_value, 3)
)
),
comb_range = if_else(
inverse_param == TRUE,
paste0(
round(parameter_upper_bound, 3),
" - ",
round(parameter_lower_bound, 3)
),
paste0(
round(parameter_lower_bound, 3),
" - ",
round(parameter_upper_bound, 3)
)
)
)
# Summarise number of studies, parameters, and contributing sources per parameter type
summary_table <- BVD %>%
group_by(parameter_type) %>%
summarise(
n_studies = n_distinct(covidence_id),
n_parameters = n(),
papers = as.character(list(unique(na.omit(article_label)))),
doi = as.character(list(unique(na.omit(doi)))),
countries = as.character(list(unique(na.omit(population_country)))),
.groups = "drop"
)
#make tidy by removing "c()"
summary_table[] <- lapply(summary_table, function(col) {
if (is.character(col)) {
col <- gsub('c\\(|\\)|"', '', col)
col <- gsub(',\\s*', '; ', col)
}
col
})
# Output saved to: Summary_tab_Bundibugyo.csv
write.csv(summary_table, "Summary_tab_Bundibugyo.csv")
# --- Main parameter table ---
BVD_clean <- BVD %>%
filter( !parameter_type == "Risk factors")%>%
mutate(
population_sample_size = ifelse((covidence_id == 2594 & (
parameter_type %in% c("Human delay - Symptom Onset/Fever to Death",
"Human delay - Admission to Care/Hospitalisation to Death"))), 28,
population_sample_size)
) %>%
select(
article_label, doi,
starts_with("parameter"),
contains("uncertainty"),
contains("bounds"),
comb_range,
contains("numerator"),
contains("denominator"),
method_r,
method_disaggregated,
method_disaggregated_by,
exponent,
starts_with("riskfactor"),
starts_with("delay"),
contains("inverse"),
contains("figure"),
population_sample_size, population_sex, population_group,
population_sample_type, population_location, population_country,
survey_date
) %>%
select(!c(
parameter_data_id, delay_start, delay_short, parameter_class,
distribution_par2_uncertainty, distribution_par1_uncertainty,
parameter_lower_bound, parameter_upper_bound,
parameter_uncertainty_lower_value, parameter_uncertainty_upper_value,
inverse_param, parameter_bounds
))
BVD_clean <- BVD_clean[, c(
"article_label", "doi", "parameter_type", "parameter_value",
"parameter_unit", "parameter_value_type",
"parameter_uncertainty_single_value", "parameter_uncertainty_singe_type",
"comb_uncertainty", "comb_uncertainty_type", "comb_range",
"cfr_ifr_numerator", "cfr_ifr_denominator",
"method_r",
"method_disaggregated", "method_disaggregated_by",
"population_sample_size", "population_sex", "population_group",
"population_sample_type", "population_location", "population_country",
"survey_date"
)]
colnames(BVD_clean) <- c(
"Article", "DOI", "Parameter", "Central value",
"Unit", "Central type",
"Single uncertainty", "Uncertainty single type",
"Uncertainty", "Uncertainty type", "Range",
"CFR Numerator", "CFR Denominator",
"R method",
"Disaggregated info available", "Disaggregation by",
"Pop sample size", "Pop gender", "Pop group", "Pop type",
"Location", "Country", "Survey date"
)
# --- Risk factor table ---
BVD_riskfactor_clean <- BVD %>%
filter(parameter_type == "Risk factors") %>%
select(
article_label, doi, parameter_type,
starts_with("riskfactor"),
population_sample_size, population_sex, population_group,
population_sample_type, population_location, population_country,
survey_date
)
BVD_riskfactor_clean <- BVD_riskfactor_clean[, c(
"article_label", "doi", "parameter_type",
"riskfactor_name", "riskfactor_outcome", "riskfactor_occupation",
"riskfactor_significant", "riskfactor_adjusted",
"population_sample_size", "population_sex", "population_group",
"population_sample_type", "population_location", "population_country",
"survey_date"
)]
colnames(BVD_riskfactor_clean) <- c(
"Article", "DOI", "Parameter",
"Risk factor name", "Outcome", "Occupation", "Significance", "Adjusted",
"Pop sample size", "Pop gender", "Pop group", "Pop type",
"Location", "Country", "Survey date"
)
# Output saved to: EBOLA_BVD_pars.csv and EBOLA_BVD_riskfactors.csv
write.csv(BVD_clean, "EBOLA_BVD_pars.csv")
write.csv(BVD_riskfactor_clean, "EBOLA_BVD_riskfactors.csv")
# Remove risk factors — not used in quantitative plots
BVD_plot <- filter(BVD, !parameter_type == "Risk factors")
# Remove Re entries with no extractable numeric value
BVD_plot <- filter(
BVD_plot,
!(parameter_type == "Reproduction number (Effective, Re)" & covidence_id == 2594)
)
plot_df <- BVD_plot %>%
# Create combined study label
mutate(
article_label_unique = make.unique(article_label),
study_label = paste0(article_label, " (", population_country, ")")
) %>%
# Replace missing uncertainty type
mutate(
parameter_uncertainty_type = case_when(
is.na(parameter_uncertainty_type) ~ "Unspecified",
TRUE ~ parameter_uncertainty_type
)
) %>%
# Replace missing value type
mutate(
parameter_value_type = case_when(
is.na(parameter_value_type) ~ "Unspecified",
parameter_value_type %in% c("Other", "", "NA") ~ "Unspecified",
TRUE ~ parameter_value_type
)
) %>%
# Order study labels by parameter value within each parameter type
group_by(parameter_type) %>%
arrange(parameter_value, .by_group = TRUE) %>%
mutate(
study_label = factor(study_label, levels = unique(study_label))
) %>%
# Fill missing units for delay parameters
mutate(
parameter_unit = case_when(
is.na(parameter_unit) & str_detect(parameter_type, "delay") ~ "(Days)",
TRUE ~ parameter_unit
)
) %>%
ungroup()
# Build facet label lookup (parameter type + unit)
unit_lookup <- BVD_plot %>%
distinct(parameter_type, parameter_unit) %>%
mutate(
facet_label = case_when(
parameter_type == "Human delay - incubation period" ~ "Incubation period\n(Days)",
parameter_type == "Attack rate" ~ "Attack rate\n(%)",
parameter_type == "Severity - case fatality rate (CFR)" ~ "CFR\n(%)",
parameter_type == "Reproduction number (Effective, Re)" ~ "R_effective",
parameter_type == "Reproduction number (Basic R0)" ~ "R_0",
parameter_type == "Mutations - substitution rate" ~ "Mutations\n(Substitutions/site/year)",
TRUE ~ paste0(parameter_type, "\n(", parameter_unit, ")")
)
) %>%
mutate(
facet_label = gsub("Human delay - ", "", facet_label),
facet_label = gsub("(inverse parameter)", "", facet_label),
facet_label = gsub("Care/Hospitalisation", "Care", facet_label),
facet_label = gsub("Recovery/non-Infectiousness", "Recovery", facet_label),
facet_label = gsub(" ()", "", facet_label),
facet_label = gsub("infectious", "Infectious", facet_label),
facet_label = gsub("time ", "Time ", facet_label)
)
plot_df <- plot_df %>%
left_join(unit_lookup, by = "parameter_type") %>%
mutate(study_label = factor(study_label, levels = rev(unique(study_label)))) %>%
group_by(facet_label, study_label) %>%
mutate(
group_id = interaction(parameter_type, study_label, parameter_value, drop = TRUE),
study_index = cur_group_id()
) %>%
ungroup()
# Colour and shape scales
uncertainty_colours <- c(
"Unspecified" = "grey70",
"95% CrI" = "#1f77b4",
"Range" = "#ff7f0e",
"95% CI" = "#2ca02c"
)
shape_values <- c(
# "Other" = 16,
"Mean" = 17,
"Median" = 15,
"Unspecified" = 1
)
plot_df$parameter_uncertainty_type <- factor(plot_df$parameter_uncertainty_type)
plot_df$parameter_value_type <- factor(plot_df$parameter_value_type)
pd <- position_dodge(width = 0.5)
# Compute orange range ribbon width
plot_width_mm <- 10 * 40
x_range <- diff(range(plot_df$study_index, na.rm = TRUE))
mm_per_data <- plot_width_mm / x_range
half_width <- (3 / mm_per_data) / 2
# Generate one plot per parameter class
plots <- list()
param_classes <- unique(plot_df$parameter_class)
dir.create("plots_png", showWarnings = FALSE)
for (i in seq_along(param_classes)) {
df_sub <- subset(plot_df, parameter_class == param_classes[i])
p <- ggplot(df_sub) +
# Orange shading for reported range
geom_rect(
aes(
ymin = parameter_lower_bound,
ymax = parameter_upper_bound,
xmin = study_index - half_width,
xmax = study_index + half_width
),
fill = "darkorange",
alpha = 0.35,
inherit.aes = FALSE
) +
# Uncertainty error bars
geom_errorbar(
aes(
x = study_index,
ymin = parameter_uncertainty_lower_value,
ymax = parameter_uncertainty_upper_value,
colour = parameter_uncertainty_type,
group = group_id
),
position = pd,
width = 0.2,
linewidth = 0.7,
na.rm = TRUE
) +
# Central estimate points
geom_point(
aes(
y = parameter_value,
x = study_index,
shape = parameter_value_type,
group = group_id
),
position = pd,
size = 2,
stroke = 1
) +
facet_wrap(
~ facet_label,
scales = "free",
nrow = ifelse(param_classes[i] == "Human delay", 3, 1)
) +
theme_bw(base_size = 12) +
labs(
x = "Study and country",
y = "Parameter value",
colour = "Uncertainty type",
shape = "Central type"
) +
scale_colour_manual(values = uncertainty_colours) +
scale_shape_manual(values = shape_values) +
coord_flip() +
scale_x_continuous(
breaks = plot_df$study_index,
labels = plot_df$study_label
) +
# scale_y_continuous(
# labels = scales::label_number(accuracy = 0.001)
# ) +
theme(
strip.text = element_text(face = "bold"),
axis.text.y = element_text(size = 8),
legend.position = "right",
panel.grid.minor = element_blank()
)
file_name <- paste0("plots_png/plot_", param_classes[i], ".png")
if (param_classes[i] == "Human delay") {
ggsave(filename = file_name, plot = p,
width = 15, height = 10, dpi = 300, scale = 0.8)
} else {
ggsave(filename = file_name, plot = p,
width = 7.5, height = 6, dpi = 300, scale = 0.8)
}
plots[[i]] <- p
}